|
 DISEASES WE INVESTIGATE DISEASES WE INVESTIGATE
Krabbé Disease
Overview
Krabbe’s disease, or Globoid Cell Leukodystrophy, is a rare, degenerative, enzyme disorder that affects cell organelles called lysosomes. It presents itself in both the central and peripheral nervous system.
- The disease is caused by a change in the genetic code (mutation) for an enzyme called galactocerebroside beta galactosidase (galactocerebrosidase, GALC).
- Although the age of onset may vary depending upon the exact mutation(s) in the gene, most cases become clinically apparent during infancy and progress rapidly to death during childhood.
- The estimated incidence in the United States is 1 in 100,000 live births.
- It has been reported in humans, dogs, sheep, cats, mice, and nonhuman primates.
- The disease was serendipitously found at the Tulane National Primate Research Center during a routine postmortem exam of an 11-week old female rhesus monkey (Macaca mulatta). The genetic defect was verified and animals that carry the defective gene also have been identified. The Tulane National Primate Research Center is the only facility to have nonhuman primates with a characterized genetic defect.
Genetic Defect
- Krabbe’s disease is caused by genetic mutation(s) in the GALC gene located on chromosome 14, which controls the generation of the lysosomal enzyme GALC.
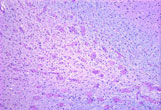
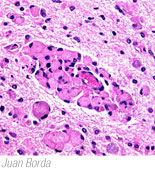
- The genetic defect causes the absence of GALC resulting in the formation of globoid cells (multinucleated macrophages), loss of oligodendroglia, and the inability to produce myelin.
- Over 70 unique gene mutations have been identified to date.
Inheritance
- Krabbe’s disease is inherited in an autosomal recessive pattern; both parents must be carriers (heterozygous, one normal and one defective GALC gene) for the trait to produce affected (homozygous, two defective GALC genes) offspring.
The Disease
- In humans, there are four clinical subtypes depending upon the age at onset of clinical signs; Infantile, Late Infantile, Juvenile and Adult. These are often referred to simply as Infantile Onset and Late Onset.
- Most cases become clinically apparent during infancy (generally 3 to 6 months of age) and rapidly progress to death during early childhood (generally by 2 years of age). Cases presenting after 10 years of age with signs and symptoms mimicking peripheral neuropathy can often result in misdiagnosis.
- Clinical and behavioral signs and symptoms include growth arrest, progressive microcephaly, severe failure to thrive, irritability, hypersensitivity to external stimuli, retardation or regression of motor skills, muscle stiffness, extension of arms and legs, clenched fingers, hypotonicity, blindness, deafness, tonic or clonic seizures, episodic fever of unknown origin, optic atrophy and/or sluggish papillary reactions to light. There is rapid and severe motor and mental deterioration in children who had previously met all developmental milestones.
- Definitive diagnosis is made by identifying the disease-causing mutation using cells from blood or tissue samples and polymerase chain reaction (PCR) techniques, or by demonstrating the absence of GALC enzymatic activity in leukocytes or cultured skin fibroblasts. Prenatal diagnosis can be made using chorionic villus samples or cultured amniotic fluid cells.
Treatment
- At present, there is no known cure for Krabbe’s disease.
- Treatment of Krabbe’s patients has been limited to supportive care and drug therapy to control irritability and spasticity.
- Transplantation of umbilical cord blood, hematopoietic stem cell transplantation and allogeneic bone marrow transplantationhave resulted in less severe signs and symptoms and/or significant delays in the onset of symptoms.
Our Research
- We use the nonhuman primate model to fully characterize the disease and disease progression using molecular, biochemical, clinical, behavioral, and pathology techniques.
- We continue to explore the biologic characterization of the disease progression and pathology in order to link clinical signs and pathology.
- We are researching new therapeutic techniques including hematopoietic stem cell transplantation, mesenchymal stem cell transplantation, neural stem cell transplantation, and Regenerative Medicine using fetal and postnatal gene transfer.
- We are presently investigating the use of recombinant virus vector-mediated Regenerative Medicine for Krabbe’s disease using fetal and postnatal gene transfer.
- Adult mesenchymal stem/progenitor cells are currently being investigated as a potential therapy for Krabbe’s disease.
- Neural stem cells isolated directly form the CNS (central nervous system) are also being investigated as a therapeutic avenue.
|
|